Зміст
- Історія вивчення
- Що відомо про поширеність?
- Яким чином успадковується хвороба?
- Роль міді в нормі і при гепатолентикулярной дегенерації
- Які зміни мозку викликають ознаки хвороби?
- Як виявляється захворювання?
- Протягом і форми хвороби
- Діагностика
- Можливості лікування
За характером морфологічних змін патологія називається гепатоцеребральной дистрофією, по авторам — хвороба Вільсона-Коновалова. Ще одне найменування — гепатолентикулярна дегенерація — уточнює і доповнює природу захворювання.
Сучасні дослідження дозволили зарахувати хвороба до уродженому порушення метаболізму сполук міді з передачею у спадщину. Виявлено відповідальний за неї ділянку мутіруючої гена. Безсумнівна зв’язок з цирозом печінки. Для хвороби Вільсона-Коновалова характерно раннє виявлення: ознаки з’являються у дітей шкільного віку і молодше.
Історія вивчення
З кінця XIX століття лікарі звертали увагу і описували неясне захворювання, якою супроводжувалося мимовільними рухами в кінцівках, м’язах тулуба, скутістю, порушенням мови, утрудненням ковтання їжі, рідше — психічними розладами зі зниженням інтелекту, розвитком слабоумства. Воно нагадувало розсіяний склероз, тому називалося «псевдосклерозом».
Англійський лікар невропатолог С. Вільсон в 1912 році вперше пов’язав симптоматику хвороби з обов’язковою наявністю циротичних змін в печінці, виявив взаємозв’язок з ураженням лентикулярных ядер головного мозку.
При цьому відзначив відсутність змін у пирамидном тракті хворих (так іменуються група ядер і нервових шляхів людини, відповідальних за руху). Значний внесок в дослідження цієї теми вніс російський вчений академік Н.В. Коновалов.
Що відомо про поширеність?
Статистика показує, що хвороба Вільсона-Коновалова виявляється у трьох осіб на 100 тис. населення. Показник вище в тих країнах, де можливі близькоспоріднені шлюби. Згідно з одним авторам — частіше хворіють чоловіки, інші стверджують, що однаково сприйнятливі як чоловіки, так і жінки.
В сім’ях, які мають носіїв патологічного гена, хворіють 25% братів і сестер. При цьому батьки можуть бути клінічно здорові. Симптоми починають проявлятися в середньому з 11 до 25 років.
Яким чином успадковується хвороба?
Тип успадкування аномального гена хвороби називається аутосомно-рецесивним. Це означає, що захворіти можуть тільки діти, які отримали відразу два мутантних гена (від матері та батька). Батьки є гомозиготними носіями.
Якщо дитина отримує ген тільки від одного з батьків, то він не хворіє, але стає гетерозиготною носієм. У нього можливі відхилення в обміні міді, але слабо виражені. Наскільки здорово буде третє покоління, залежить від зустрічі з аналогічно зміненою структурою хромосом дружини або чоловіка.
Відомий ген хвороби — АТР7В — він забезпечує синтез транспортуючого мідь білка (церулоплазміну). Знаходиться на довгому плечі хромосоми №13, на ділянці з кодом 13q14-q21.

Мутації пов’язані з заміною послідовності включення амінокислот
Виявлено різні види мутацій у пацієнтів в Китаї та країнах Заходу. Причини порушень залишаються неясними. У 10% хворих взагалі не виявлено генні зміни. Встановлено, що у прояві захворювання важливе значення належить факторам, що вражає печінку. До них належать перенесені інфекційні хвороби (вірусний гепатит), інтоксикація.
Роль міді в нормі і при гепатолентикулярной дегенерації
Здорова людина отримує щодня з їжею 2 мг міді, з них засвоюється лише 1/3 частина, але і цього цілком вистачає для забезпечення потреби організму.
Мідь необхідна печінки:
- для синтезу білків і ферментів, а значить для росту, фізичного і розумового розвитку;
- утворення гемоглобіну, транспорту заліза;
- синтезу еритроцитів і лейкоцитів;
- забезпечення еластичності стінок судин за рахунок участі в синтезі колагену;
- стимуляції гормональної активності гіпофіза та інсуліну;
- вироблення клітин імунітету;
- освіти антиоксидантів, запобігання раннього старіння.
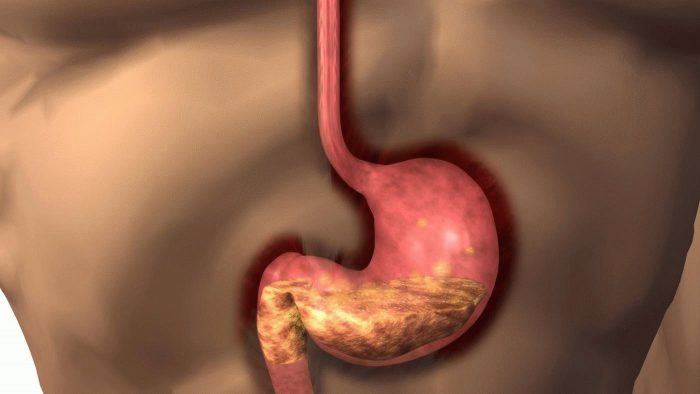
 Ознаки хвороби печінки
Ознаки хвороби печінки
Потрапляючи їх тонкого кишечника в кров, по ворітній вені молекули міді доставляються в печінкові клітини. Тут частина зв’язується з білком церулоплазмином і повертається у кров’яне русло, інша (зайва) — переходить у жовч і видаляється з організму. Одна молекула церулоплазміну включає 8 атомів міді.
У патогенезі хвороби Вільсона-Коновалова порушуються обидві функції. Церулоплазмин руйнується. Кров хворого переповнюється значною кількістю незв’язаної міді. Вона відкладається в різних органах: печінці, головному мозку, нирках, рогівці очей, переходить в сечу.
В гепатоцитах печінки, як відповідна реакція на впровадження міді утворюється запалення, що переходить у вузловій цироз. В нирковій тканині страждають проксимальні канальці. Пошкодження головного мозку виражається в ураженні базальних гангліїв, ядер мозочка і чорної субстанції.
В тканинах ока мідь відкладається навколо рогової оболонки і утворює видимий кільце Кайзера-Флейшера. Переважне відкладення міді в одному з органів залежить від віку. Для дітей типово ураження печінки. Після 20 років основні руйнування локалізуються в головному мозку.
Які зміни мозку викликають ознаки хвороби?
Гепатоцеребральная дистрофія викликає в тканинах головного мозку розм’якшення декількох ядерних утворень (лентикулярного, хвостатого зубчастого, подбугорных), мозочка, глибинних шарів коркового шару. В них утворюються дрібні кісти.
Характерні токсичні зміни розрізняють:
- судинні атонія дрібних судин сприяє застійним явищам крові, набряку навколишнього тканини, загибелі клітин, крововиливів з виділенням гемосидерину;
- клітинні — дистрофії піддається макроглия нейронів, можливе формування глії Альцгеймера, з часом клітини гинуть.
Встановлено, що клітинні порушення більш поширені при пізньому початку хвороби і повільному плині. У печінці атрофічний процес призводить до загибелі гепатоцитів, цілих часточок з утворенням некротизованих вузлів. Між ними залишаються осередки відновлення, нормальні клітини.
Утворюються нові судини, які формують зв’язки між гілками ворітної вени печінки і руслом нижньої порожнистої
Як виявляється захворювання?
Для гепатоцеребральной дистрофії характерно початок у дитячому віці або до 25 років, подальший розвиток приймає хронічний перебіг з прогресуванням клінічних проявів. Часто до появи неврологічних симптомів хвороби Вільсона-Коновалова виявляють розлади функцій печінки, кишечника.
У пацієнтів виникають тупі болі в підребер’ї праворуч, жовтяниця, діарея або запор. Рідше розвивається гепатолієнальний синдром. Серед неврологічних ознак:
- гіперкінези (мимовільні рухи в руках та ногах, тремор), схожі на кривляння;
- м’язові спазми (ригідність) і судоми;
- паралічі;
- епілептиформні напади;
- порушена мова;
- слинотеча;
- розлади поведінки, психіки.
На відміну від ішемічних порушень і крововиливів немає порушень чутливості.
Серед інших симптомів хвороби, що вказують на ураження внутрішніх органів:
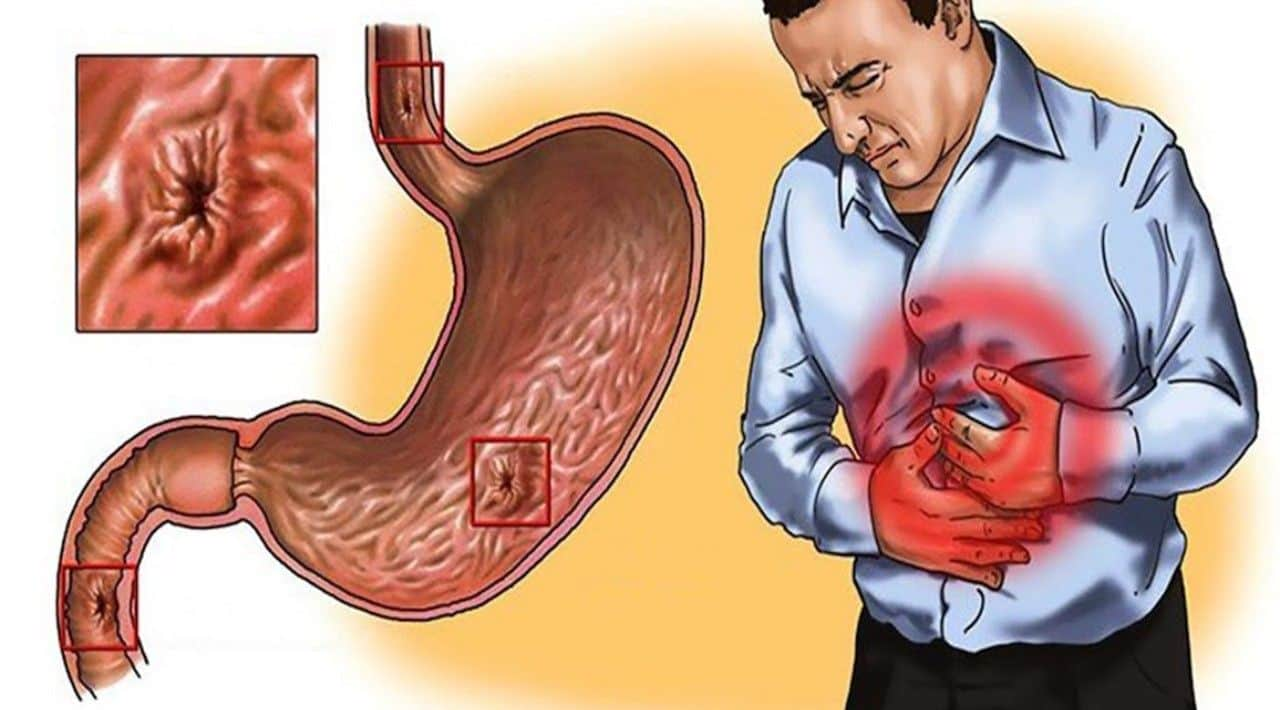
- кільце Кайзера-Флейшера — зеленуваті відкладення міді по колу рогової оболонки, з’являються на пізній стадії хвороби;
- пігментні плями на шкірі обличчя і тулуба жовтувато-коричневого кольору;
- ознаки геморагічних порушень — часті носові кровотечі, кровоточивість ясен, мармуровий відтінок шкіри, цианотичность губ, пальців;
- болі в суглобах;
- підвищена ламкість кісток, переломи через остеопорозу;
- профузні виділення поту.
Симптоми патологічних порушень в печінці виявляються в 1/3 випадків, частіше їх можна виявити в аналізах крові з підвищення рівня трансаміназ, білірубіну, зниження тромбоцитів і лейкоцитів, гемолітичної анемії. Печінка значно збільшена.
Клінічні обстеження підтверджують активний гепатит з переходом в цироз
Протягом і форми хвороби
За характером перебігу хвороби розрізняють гостру і хронічну форми. Гостра — типова для раннього дитячого віку, відрізняється блискавичним розвитком, швидким летальним результатом навіть при лікуванні. Хронічна форма протікає повільно, з поступовим обважненням неврологічної симптоматики.
Залежно від переважного ураження органів прийнято виділяти п’ять різновидів захворювання. Дрожательно-ригидная — зустрічається найбільш часто, починається в юнацькому віці, протікає повільно з ремісіями і загостреннями.
Стан погіршується раптово, трохи підвищується температура тіла, в руках визначається поєднання ригідності м’язів і ритмічних дрожательных посмикувань по 2-8 рухів в секунду. Прояви посилюються при м’язовому напрузі, заворушеннях, а в спокійному стані, уві сні зникають.
Можливо порушення ковтання і мови, насильницькі рухи (атетоидные, аналогічні хореї). Пацієнти живуть в середньому 6 років.
Дрожательная — виникає до 20 років, відрізняється повільним плином. В клініці хвороби переважає тремтіння рук. М’язова ригідність з’являється багато років потому. Характерна втрата міміки обличчя, повільна мова без емоцій, іноді зниження тонусу м’язів. Значно страждає психіка людини, можливі спалахи агресії, епілептиформні припадки. Тривалість життя пацієнтів до 15 років і більше.
Черевна — найбільш важкий варіант перебігу хвороби Вільсона-Коновалова у дітей. Ураження печінки з вираженою недостатністю призводить до раннього смертельного результату. Неврологічна симптоматика не встигає розвинутися. Тривалість захворювання не перевищує 5 років, дитина може загинути через кілька місяців.
Ригидно-аритмогиперкинетическая — інша назва — рання форма. Починається в дитинстві, характерно швидко прогресуючий перебіг. У клініці проявляється значною м’язовою ригідністю зі стійкими контрактурами, сповільненістю і труднощами пересування, порушеною промовою і ковтанням, частими хореоатетоидными рухами. Інтелект знижується помірно. Хвороба триває протягом двох-трьох років до загибелі дитини.
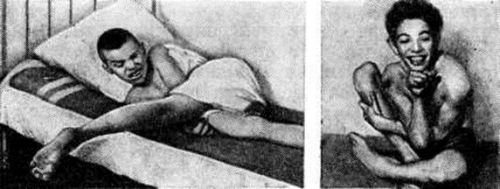
У пацієнтів виникає судомний плач або сміх
Экстрапирамидно-коркова — рідкісна форма перебігу хвороби. До характерних ознак додаються парези і паралічі, важкі напади епілепсії, розвиток повного недоумства. У корі півкуль мозку утворюються великі вогнища розм’якшення. Середня тривалість — 6-8 років. Неминучий летальний результат.
Сама рання смертність у дітей з черевної формою. У половині випадків хвороби спостерігається масивний некроз печінки, гемолітичні порушення, шлунково-кишкова кровотеча (наслідки портальної гіпертензії). Від неврологічних змін пацієнти без лікування гинуть через 5-15 років.
Діагностика
Діагноз ставиться на підставі клінічної симптоматики, дані про захворюваність в сім’ях батьків, укладення генетичних досліджень. У спеціалізованих неврологічних відділеннях перевіряють:
- кількість міді в сироватці крові;
- концентрацію церулоплазміну (обидва показника при хворобі знижуються);
- виділення міді з сечею (зростає);
- огляд окуліста з допомогою щілинної лампи дозволяє виявити кільця Кайзера-Флейшера, іноді замість повного кільця виявляють «уламки».
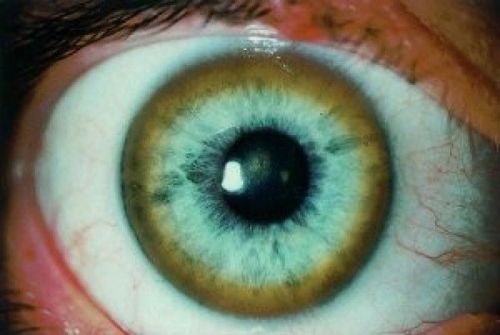
На фото з щілинної лампи добре видно зеленувате кільце, воно є не у всіх пацієнтів
Мають значення біохімічні проби печінки, зростання трансаміназ, лужної фосфатази, білірубіну, тимолової проби, зміна білкових фракцій, виявлення знижених показників згортання.
Для загального аналізу крові типово зниження еритроцитів і тромбоцитів, лейкопенія. Ниркові канальцевые зміни проявляються виявленням в сечі глюкози, солей фосфорної кислоти, уратів, білка.
Комп’ютерна та магнітнорезонансна томографія черепа виявляє зміни мозку до появи характерних неврологічних симптомів: збільшення (розширення) шлуночків, вогнищеві порушення в зоні таламуса, шкаралупи і блідої кулі. У складних випадках проводять біопсію печінки, визначають вміст міді в печінці.
Для генетичних досліджень хвороби Вільсона-Коновалова використовують ДНК-маркери, які володіють високою точністю. У диференціальній діагностиці враховують, що церулоплазмин знижується при гепатитах, перенесеному голодуванні.
Можливості лікування
Наведені терміни тривалості життя пацієнтів при сучасному рівні медицини вдається значно збільшити за допомогою раннього початку лікування хвороби Вільсона-Коновалова та використання всіх можливих засобів.
Харчування пацієнта повинно проводитися згідно з дієтичного стола №5а (для захворювань печінки). Виключаються всі гострі страви, смажені і жирні. Одночасно для обмеження надходження міді з їжею категорично забороняються: вироби і натуральні продукти, що містять шоколад, горіхи, кава, м’ясо печінки, раків і кальмарів, сухофрукти, бобові, крупи з цільної пшениці.
Основний напрямок — забезпечення виведення надлишків міді. Головним препаратом вважається D-пеніциламін. Препарат призначається з невеликої дози за схемою, потім вона збільшується. Необхідно враховувати довічний прийом ліків і його негативні властивості.
Фахівці звертають увагу на низьку якість вітчизняного Пеніциламіну, його токсичність. У деяких пацієнтів з гепатолентикулярной дегенерацією побічні явища полягають у дерматитах, анемії.
Препарат утворює з міддю комплекси, які переходять у сечу і виділяються з організму, крім того, здатний пригнічувати активність внутрішньоклітинної міді
Інший напрямок — терапія солями цинку (сульфат, оксид). Застосування комбінацій D-пеніциламін з препаратами цинку дозволяє проводити терапію низькими дозами та уникати негативних наслідків. Якщо захворювання виявлено до початку клінічних проявів, то рекомендуються тільки препарати цинку. Доповненням є використання Унітіолу.
В обов’язковому порядку пацієнту призначаються гепатопротекторні засоби: вітаміни, Есенціале. Для підтримки провідності в нервових волокнах необхідні вітаміни групи В. Запропоновано спосіб біо-гемоперфузії крові з живими ізольованими клітинами печінки і селезінки. Методику називають «допоміжної печінкою».
Вона проводиться в спеціалізованих центрах при безуспішної терапії для підтримки організму пацієнта до операції трансплантації печінки. Пересадка здорової печінки донора допомагає вирішити безліч проблем лікування.
Сама операція потребує підготовки хворого, обліку ризиків в зв’язку зі зниженою згортанням крові. Це найбільш радикальний метод лікування форм хвороби з переважним ураженням печінки.
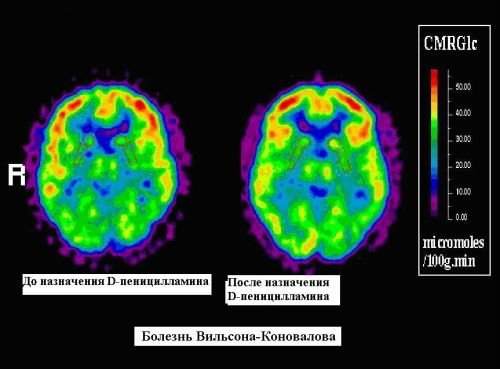
Магніто-резонансне обстеження застосовують в процесі контролю за лікуванням
Ефективність лікування значно поліпшується при ранній діагностиці. Вдається досягти зменшення проявів. Пацієнти залишаються соціально адаптованими людьми: здатні повністю до самообслуговування, навчаються, працюють за фахом, створюють сім’ю. Є спостереження за молодими жінками з хворобою Вільсона-Коновалова, выносившими вагітність і родившими здорових дітей.
Раз ефект лікування хвороби залежить від раннього виявлення і початку, то сім’ям, в яких є дитина з гепатолентикулярной дегенерацією, необхідно обстежити його братів і сестер із застосуванням сучасних молекулярно-генетичних методик. Знання майбутніх батьків про своє носійстві запобіжить народження хворої дитини.